

Opti-Protein Ultra Marker
| Cat. No. | G623 | ||||||||
| Name | Opti-Protein Ultra Marker | ||||||||
| Category | DNA & Protein Ladders | ||||||||
| Description |
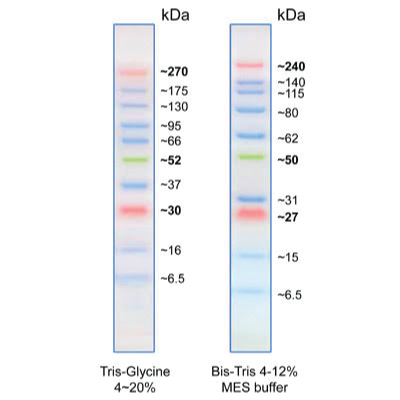
With enhanced performance for high molecular weight proteins, Opti-Protein Ultra Marker consists of 10 pre-stained proteins that resolve into sharp, tight bands covering a wide range of molecular weights from 6.5 to 270 kDa. This protein ladder allows you to monitor protein separation during electrophoresis, estimate molecular weight of the protein of interest, and evaluate western blot transfer efficiency. Proteins are covalently coupled with a blue chromophore except for three reference bands (two orange bands at 30 kDa and 270 kDa and one green band at 52 kDa) when separated on gel. Features:
|
||||||||
| Application |
|
||||||||
| Storage Condition |
Store @ 4°C for 3 months or -20°C for 24 months. |
||||||||
| Material Citation | If use of this material results in a scientific publication, please cite the material in the following manner: Applied Biological Materials Inc, Cat. No. G623 | ||||||||
| Why do the molecular weights of protein markers appear different in MOPS, MES, and Tris-Glycine buffers? | |
|
1. Protein shape and migration change in different buffers: 2. Prestained markers behave differently than unstained proteins: 3. Molecular weights are calibrated for accuracy: |
| Why I do have high background? | |
|
High background is often caused by insufficient blocking, high antibody concentration, or non-specific binding. To reduce it: Blocking: Block for at least 30 minutes using 5% non-fat dry milk, 3% BSA, or normal serum. These can also be included in antibody buffers. For phospho-specific antibodies, avoid milk (it contains casein, a phosphoprotein) and use BSA instead. Antibody Optimization: Use the lowest effective antibody concentration. Incubate longer at lower concentrations to improve specificity. Incubation Conditions: Perform antibody incubations at 4°C to reduce non-specific interactions. Secondary Antibody Issues: Secondary antibodies may bind non-specifically or react with blocking agents. Run a secondary-only control to check for this. Washing: Increase the number and duration of washes, and include Tween-20 in your buffers to help remove unbound antibodies. Membrane Type & Care: Nitrocellulose typically produces less background than PVDF. Always keep the membrane moist during all steps, as drying can increase background. |
| Why do I have multiple bands? | |
|
1. Cell Line Variation: Frequently passaged cell lines may show altered protein expression. Compare results with the original, low-passage line by running both samples side by side. 2. Post-Translational Modifications (PTMs): Proteins may be phosphorylated, glycosylated, acetylated, etc., causing shifts in size. Use enzymatic treatments (e.g., phosphatases, deglycosylases) to confirm. 3. Protein Degradation: Low molecular weight bands may be degradation products. Use protease inhibitors in your lysis buffer to prevent this. 4. Alternative Splicing or Isoforms: The antibody may detect splice variants or related proteins with shared epitopes. Check the literature and perform a BLAST search for related sequences. 5. High Antibody Concentration: Too much primary or secondary antibody can cause non-specific bands. Dilute your antibodies and run a secondary-only control to check. 6. Antibody Purity: Unpurified antibodies may bind non-specifically. Use affinity-purified antibodies for better specificity. 7. Non-specific Binding: Confirm specificity using a blocking peptide—specific bands should disappear when blocked. 8. Protein Multimers: Your target may form dimers or higher-order structures. Try boiling in SDS sample buffer for 10 minutes to disrupt multimers. |
| Why are there uneven white “spots” on the blot? | |
|
Air bubbles were trapped against the membrane during transfer or the antibody is not evenly spread on the membrane. Make sure you remove bubbles when preparing the gel for transfer. Incubate antibodies under agitation.
|
| Why do I have black dots on the blot? | |
|
The antibodies are binding to the blocking agent. Filter the blocking agent.
|
| White bands on a black blot (negative of expected blot)? | |
|
Too much primary and/or too much secondary antibody. Dilute the antibodies more.
|
| MW marker lane is black? | |
|
The antibody is reacting with the MW marker.
Add a blank lane between the MW marker and the first sample lane.
|
| The band of interest is very low/high on the blot? | |
|
It is due to separation is not efficient.
Change the gel percentage: a higher percentage for small protein, lower percentage for large proteins.
|
| Smile effect of the bands? | |
|
Two possibilities:
1. Migration was too fast. 2. Migration was too hot (changing the pH and altering the migration). Slow down the migration or run the gel in the cold room or on ice. |
| Uneven band size in lanes probed for the same protein? | |
|
Gel has set too quickly while casting and the acrylamide percentage is not even along the lanes.
Review the recipe of the gel and the addition of TEMED to the gels, add a little 0.1% SDS in water to the top of the migrating gel while it sets to stop it from drying.
|
| Uneven staining of the gel? | |
|
Two possibilities:
1. Contamination from bacteria. 2. Not enough antibody. |
| Is it pre-stain or un-stain? | |
|
All the OptiProtein marker are pre-stained and ready to load directly on the gel.
|
| I do not see any bands on my blot. Are there any suggestions? | |
|
The best possible control for the antibody is to transfect 293 cells with your gene of interest, produce lysate from these cells, and then use them for western blot. For other lysates from cells that do express the protein of interest naturally, the expression level may not be strong enough for detection and it is best to work with 293 cells first to prove if the antibody is functional in your hands. Also, removing all detergent and incubating the primary antibody overnight will help produce stronger band signals.
|
| How many micrograms of protein are in each band of the marker? | |
|
We have about 0.1 - 0.2 mg per protein band for each 500 µl vial of the prestained protein ladder.
|
| Do the protein markers work against native gels? | |
|
Our protein markers can run though a native gel. However, all our protein markers contain denatured proteins and thus, cannot be equally compared to native proteins in the native gel setting.
|
| How much of the should I load onto the gel? | |
|
It is recommended to gently mix the ladder by pipetting up and down a few times, then load 5 μl directly into the well of the gel.
|